THE ROLE OF GENETICS
Cystic fibrosis (CF) is a genetic disease caused by specific changes (mutations) in our genetic material. Our genetic material – DNA – is a combination of genes inherited from our parents. The information is stored in DNA as a code made up of different “letters”. Any change in the “letters” could cause defects in the final gene product – the protein.
CF is caused by mutations in the CFTR gene1,2. The CFTR gene codes for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Each person has two copies of the gene, one inherited from each parent. CF is a recessive disease, meaning that it manifests only if a person has two copies that contain mutations (i.e., are defective). A person with one functional copy of CFTR and one copy containing a mutation does not have CF, but is a carrier and can pass the defective copy on to their children. CF is not linked to sex, so a child will have the same chance of inheriting CF whether it is a boy or a girl. However, there is a discrepancy between adult women and men with CF – men usually live longer3.
How Likely Will My Child Have Cystic Fibrosis?
To have CF, both parents must be carriers of the disease or have CF. In the Caucasian population, about 1 in 25 is a carrier of CF4. As carriers do not have any symptoms of CF, they usually do not know that they are carriers until they have a child with CF. Statistically speaking, two parents of Caucasian origin with no information about their carrier status have a 0.04 % chance that their child will have CF. If two carriers have a child, the chances are:
- 25 % that the child will have CF
- 50 % that the child will be a carrier of CF without having CF themselves
- 25 % that the child will not be a carrier or have CF
If one of the parents has CF, the child will have CF with a 50 % chance if the other parent is a carrier. If both parents have CF, all their children will inherit CF.
CFTR Mutations
So far, there have been more than 2000 described variants of the CFTR gene, but not all of them cause CF5. Only a much smaller number of mutations is commonly present in people with CF. Each person with CF has two mutations – these can be the same or different. The majority of people with CF have at least one copy of the most common mutation – F508del6. Different types of mutations in the CFTR gene cause various effects on the protein. First, let us see how a functional CFTR protein is made.
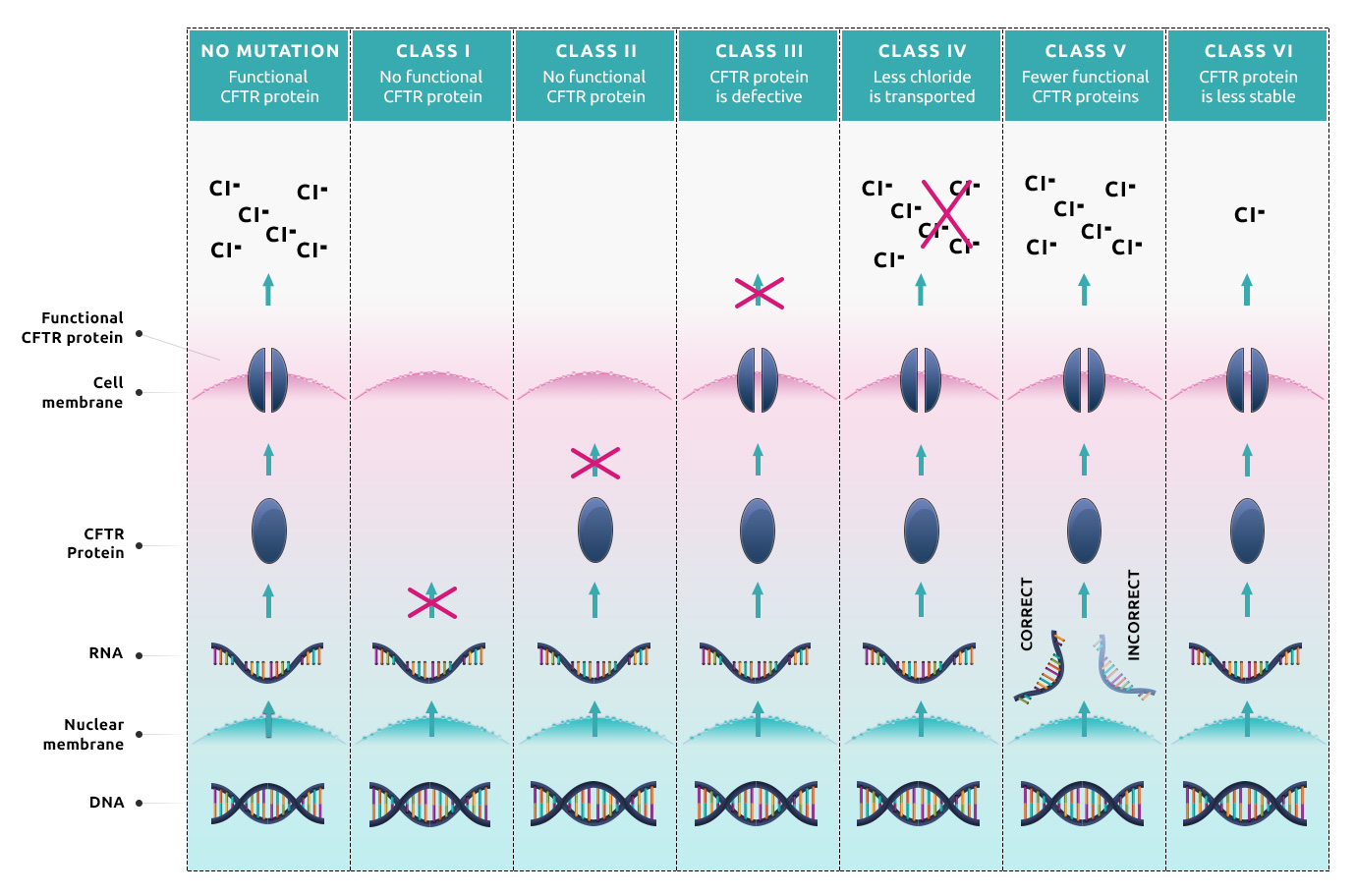
Instructions for the CFTR protein are coded in the DNA located in the cell nucleus. To make the protein, first, the instructions must be transcribed into RNA, which then serves as a template for protein synthesis. The protein synthesis stops once the machinery comes across a termination signal. The synthesized protein needs to fold correctly because its spatial structure is important for proper function. Then the mature protein moves through the cell to get to the cell surface, where it serves in the membrane as a channel to transport ions from the inside of the cell to the outside.
The most recent classification divides the various CFTR mutations into several categories6,7:
- Class I: These mutations cause no functional CFTR protein to be formed. This
group can be further divided into classes A and B.
- Class IA: These mutations cause the absence of the template RNA (e.g., large parts are missing) and so far, they cannot be treated with therapeutics. Examples include dele2,3(21kb).
- Class IB: These are nonsense variants, where either the template RNA is degraded, or the protein synthesis ends prematurely, which leads to the absence of the protein. Examples include G542X, W1282X, or R553X.
- Class II: These mutations cause CFTR trafficking defects. No CFTR protein is in the cell membrane, because it was degraded before reaching the cell surface. Examples include F508del, S549T, A559T, N1303K, or I507del.
- Class III: These mutations cause defective channel regulation. The protein is located at the proper place and in normal amounts, but it does not open correctly when stimulated. Examples include G551D, G551S, G1244E, or G1349D.
- Class IV: These mutations result in decreased channel conductance. This causes fewer chloride ions being transported to the outside of the cell when the channel is open. This mutant version of CFTR has residual activity. Examples include R334W, R347P, or R117H.
- Class V: These mutations cause a substantial reduction in the RNA or protein level, resulting in a reduced number of CFTR protein in the cell membrane. Fewer functional proteins can transport fewer ions so this defect also needs to be treated. Examples include 3272-26A>G, 2789+5G>A, or 3849+10kbC>T.
- Class VI: These mutations result in a reduction of protein stability, where the protein is degraded rapidly. Examples include c.120del23 or rF508del (r = rescued).
When you have CF, it is important to know what defects your genetic mutations cause, because they require different therapeutic approaches. Matching therapies to specific types of genetic mutations – theratyping – is essential so that people with various CF-causing mutations can truly benefit from the progress in CF research (see Personalized treatment).
References
- 1 - B. Kerem et al., “Identification of the cystic fibrosis gene: genetic analysis.,” Science, vol. 245, no. 4922, pp. 1073–80, Sep. 1989.
- 2 - J. M. Rommens et al., “Identification of the cystic fibrosis gene: chromosome walking and jumping.,” Science, vol. 245, no. 4922, pp. 1059–65, Sep. 1989.
- 3 - C. L. Harness-Brumley, A. C. Elliott, D. B. Rosenbluth, D. Raghavan, and R. Jain, “Gender differences in outcomes of patients with cystic fibrosis,” J. Women’s Heal., vol. 23, no. 12, pp. 1012–1020, Dec. 2014.
- 4 - J. Massie and M. B. Delatycki, “Cystic fibrosis carrier screening,” Paediatric Respiratory Reviews, vol. 14, no. 4. pp. 270–275, Dec-2013.
- 5 - http://www.genet.sickkids.on.ca/StatisticsPage.html
- 6 - S. V. N. Pereira, J. D. Ribeiro, A. F. Ribeiro, C. S. Bertuzzo, and F. A. L. Marson, “Novel, rare and common pathogenic variants in the CFTR gene screened by high-throughput sequencing technology and predicted by in silico tools,” Sci. Rep., vol. 9, no. 1, Dec. 2019.
- 7 - J. S. Elborn, “Cystic fibrosis - J Stewart Efborn,” Lancet, vol. 388, pp. 2519–2531, 2016.